A fragile cleanup system sitting on the surface of brain cells may help explain one of Alzheimer’s disease’s oldest mysteries. It may reveal how ordinary tau protein first turns into the twisted filaments tied to memory loss and cognitive decline.
That is the central finding from a Columbia University team that traced the earliest stages of tau damage to a neuron-specific protein disposal system called the neuroproteasome. When that system was disrupted, tau rapidly misfolded into paired helical filaments. This is the same broad kind of abnormal structure seen in the brains of people with Alzheimer’s disease.
The work points to a possible starting point for tau pathology. Additionally, it connects two of the disease’s biggest risk factors: aging and the APOE4 gene variant.
“These prior studies could not capture how tau misfolds in the first place in Alzheimer’s disease but understanding how tau aggregation begins is critical if we want to create therapies that prevent neurodegeneration before it starts,” says the study’s senior author, Kapil Ramachandran, assistant professor of neurological sciences at Columbia University.
Tau normally helps stabilize neurons. In Alzheimer’s, it becomes misshapen and collects into tangles. Those tangles are closely tied to worsening cognitive symptoms. Yet, researchers have struggled to explain how normal tau begins that shift in the first place. This is especially true because it does not misfold in standard animal models the way it does in people.
Ramachandran’s lab had previously identified the neuroproteasome as an extra disposal system that spans a neuron’s outer membrane. It appears to destroy newborn proteins that are especially vulnerable to misfolding.
“In our new study, we wanted to see what would happen if we blocked the neuroproteasome,” Ramachandran says. “We had to build a set of molecular tools to make that happen. And that’s when we found the tau filaments.”
To do that, the team created membrane-impermeable inhibitors designed to block the neuroproteasome. These did so without shutting down the cell’s internal proteasomes. That distinction mattered. This is because standard proteasome inhibitors affect the whole cell and trigger broader cleanup responses. Those responses can mask what is happening specifically at the neuronal surface.
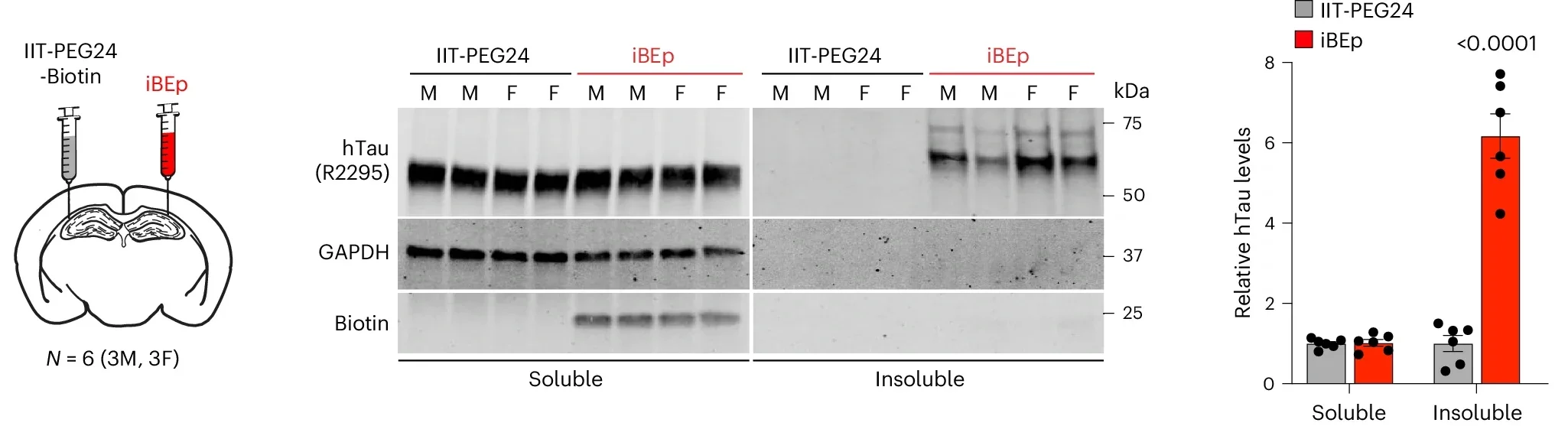
With the new inhibitors, the researchers found that blocking the neuroproteasome in primary mouse neurons caused endogenous tau to shift into a sarkosyl-insoluble form. This is a biochemical signature associated with aggregation. The same thing happened in neurons from mice carrying human tau at normal levels, without mutations or overexpression.
The effect did not appear when tau was absent. It also did not appear with inactive control compounds, strengthening the case that the neuroproteasome itself was driving the change.

The team then asked whether these newly formed aggregates resembled the tau structures seen in Alzheimer’s.
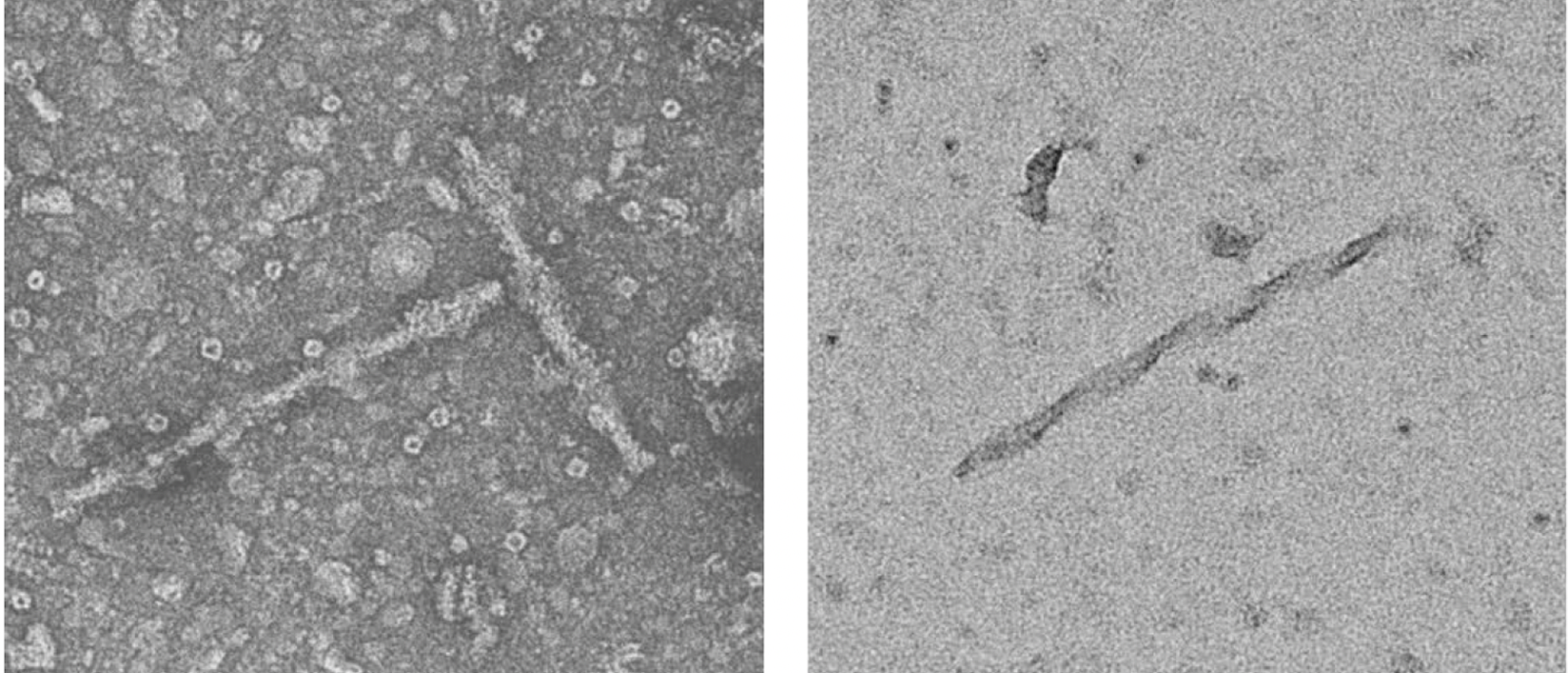
In mice, after injecting the inhibitor into the hippocampus, a brain region heavily affected in Alzheimer’s, the researchers found substantial amounts of insoluble tau. Electron microscopy showed twisted filaments resembling paired helical filaments. Moreover, immunogold labeling confirmed those filaments contained tau.
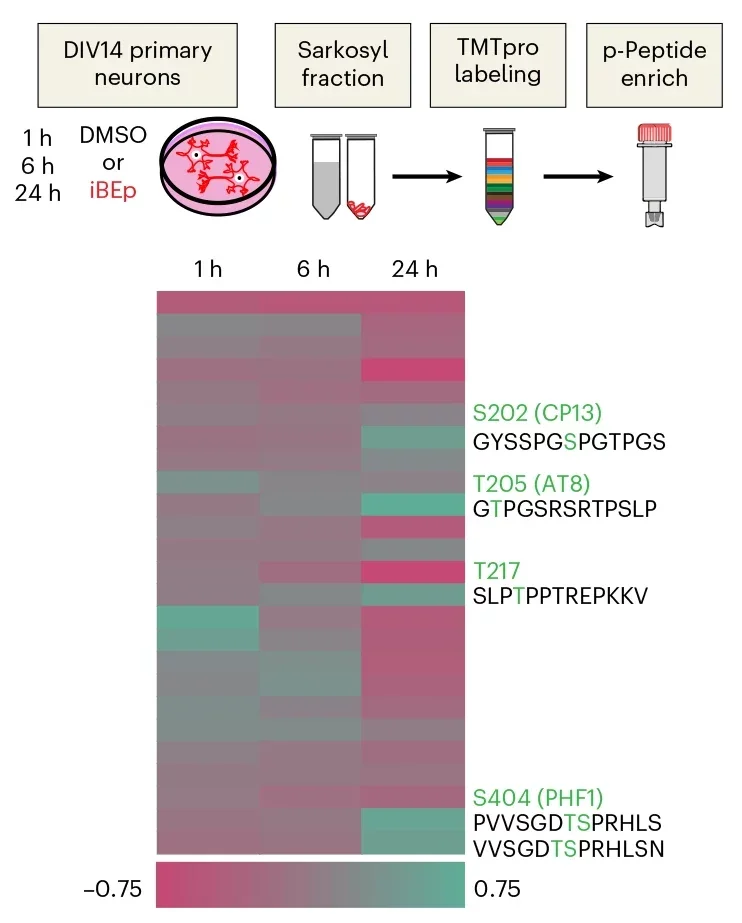
The structures also carried other Alzheimer’s-like traits. Tau showed increased phosphorylation at several sites that match known markers in human Alzheimer’s brain tissue. These sites include those corresponding to human Ser202, Thr205, Thr217 and Ser404. In brain sections, the group detected elevated AT8 staining, a standard marker of phosphorylated tau. There was also Thioflavin S-positive flame-like and thread-like inclusions, both associated with aggregated protein assemblies.
Taken together, the results suggest that selective neuroproteasome disruption is enough to trigger the de novo formation of Alzheimer’s-relevant tau aggregates from normal endogenous tau.
That matters because most earlier work has relied on introducing preexisting tau tangles from patient brains into animals. This study instead focused on how the process may begin.
The findings became more striking when the team looked at ApoE, a protein already central to Alzheimer’s risk. APOE4 is known to sharply raise the odds of developing the disease, while APOE2 is considered protective.
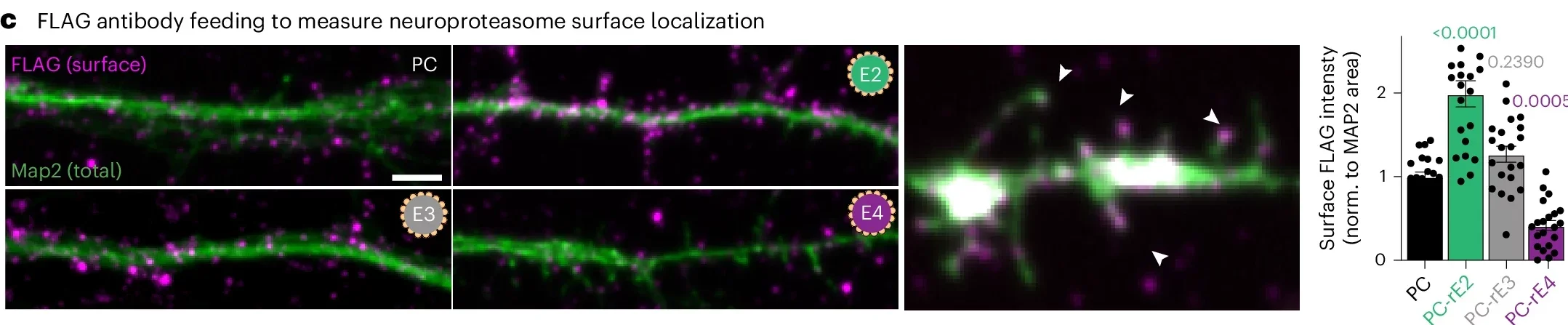
The researchers found that ApoE and its receptor Lrp1 interact with the neuroproteasome. In humanized ApoE mouse models, neuroproteasome levels at the cell surface were strongly reduced in ApoE4 mice compared with ApoE3 and ApoE2 mice. ApoE2 had the opposite effect. It increased neuroproteasome localization.
The same pattern held in cultured neurons treated with extracellular lipidated ApoE particles. Lipidated ApoE4 reduced surface neuroproteasomes, while lipidated ApoE2 increased them. Delipidated ApoE did not have the same effect.
That shift appears to change how close neurons are to a tipping point. In neurons carrying both human tau and human ApoE variants, ApoE4 cells began accumulating insoluble tau after only about 20% of neuroproteasome activity was lost. ApoE3 neurons crossed that line after about 60% loss. Furthermore, ApoE2 neurons not until roughly 85% was inhibited.
In other words, ApoE4 did not cause tangles by itself, but it left neurons with far less reserve.
“The links between tau filament formation and APOE variants and aging, Alzheimer’s greatest risk factors, suggest we may have found a mechanism to explain how an important aspect of the disease gets started,” Ramachandran says. “Our hope now is that our findings will lead to the development of therapies that prevent tau tangles from forming in the first place.”
Aging, the biggest overall risk factor for Alzheimer’s, also appeared to erode this protective machinery.
In wild-type mice, membrane-localized neuroproteasomes declined progressively with age, beginning around 12 months. Human brain tissue showed a related pattern. People with two copies of APOE4 had significantly lower neuroproteasome levels than APOE3 carriers, and reductions were especially pronounced in brain regions with heavy tau pathology.
The human tissue analysis compared Brodmann area 7, a region with substantial tau tangles and neurodegeneration, with Brodmann area 4, which has much less tau pathology. Neuroproteasome levels were lower in APOE4 carriers regardless of region. However, the drop was greater in the more affected area.
That does not prove the neuroproteasome is the sole cause of Alzheimer’s, and the study does not recreate the slow, widespread course of the disease in people. The experiments relied on acute disruption of the system, not the chronic buildup that unfolds over years. The authors also note that ApoE4’s reduction of neuroproteasome levels is not sufficient on its own. A second hit, such as aging or another stressor, may be needed to push neurons past the threshold where tau begins to aggregate.
Still, the work offers a concrete mechanism tying together tau, APOE genotype and age-related vulnerability.
That could help redirect treatment strategies.
Amyloid-clearing drugs have shown modest benefits, but many researchers believe tau may be more tightly linked to the course of cognitive decline. If the neuroproteasome helps determine whether tau stays soluble or begins forming filaments, then preserving that system, or counteracting the effect of ApoE4 on it, could become a new therapeutic goal.
The study also raises broader questions. The researchers say future work will need to determine exactly how ApoE influences neuroproteasome localization. Another question is why neurons do not appear to launch standard protective responses when this surface system is impaired. Moreover, they must find out how this pathway relates to later events such as tau spreading, neurodegeneration and disease progression.
For now, the study offers something Alzheimer’s research has long lacked: a plausible glimpse of how normal tau may first take a dangerous turn.
The findings suggest that future Alzheimer’s therapies may need to act earlier than today’s treatments, before tau tangles fully form.
By identifying the neuroproteasome as a possible control point, the research opens a path toward drugs that preserve this disposal system, strengthen a neuron’s resistance to stress, or blunt the harmful effect of APOE4.
It also gives researchers a new way to study how tau aggregation begins, which could improve screening for preventive therapies aimed at slowing disease before major memory symptoms appear.
Research findings are available online in the journal Nature Neuroscience.
The original story “Columbia University researchers discover new clues to Alzheimer’s origins” is published in The Brighter Side of News.
Like these kind of feel good stories? Get The Brighter Side of News’ newsletter.
The post Columbia University researchers discover new clues to Alzheimer’s origins appeared first on The Brighter Side of News.

